
ELTD1 Activation Induces an Endothelial-EMT Transition to a Myofibroblast Phenotype

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
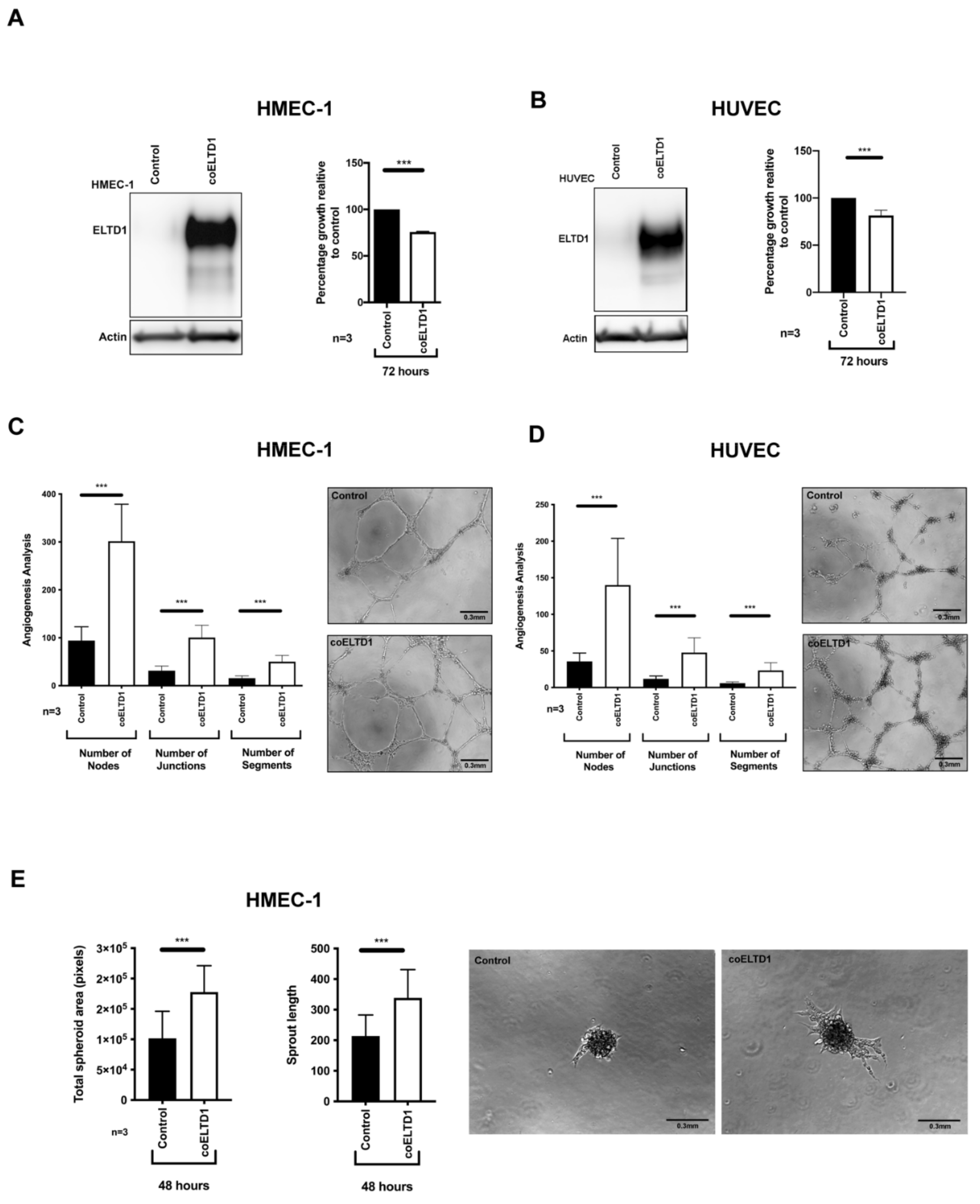
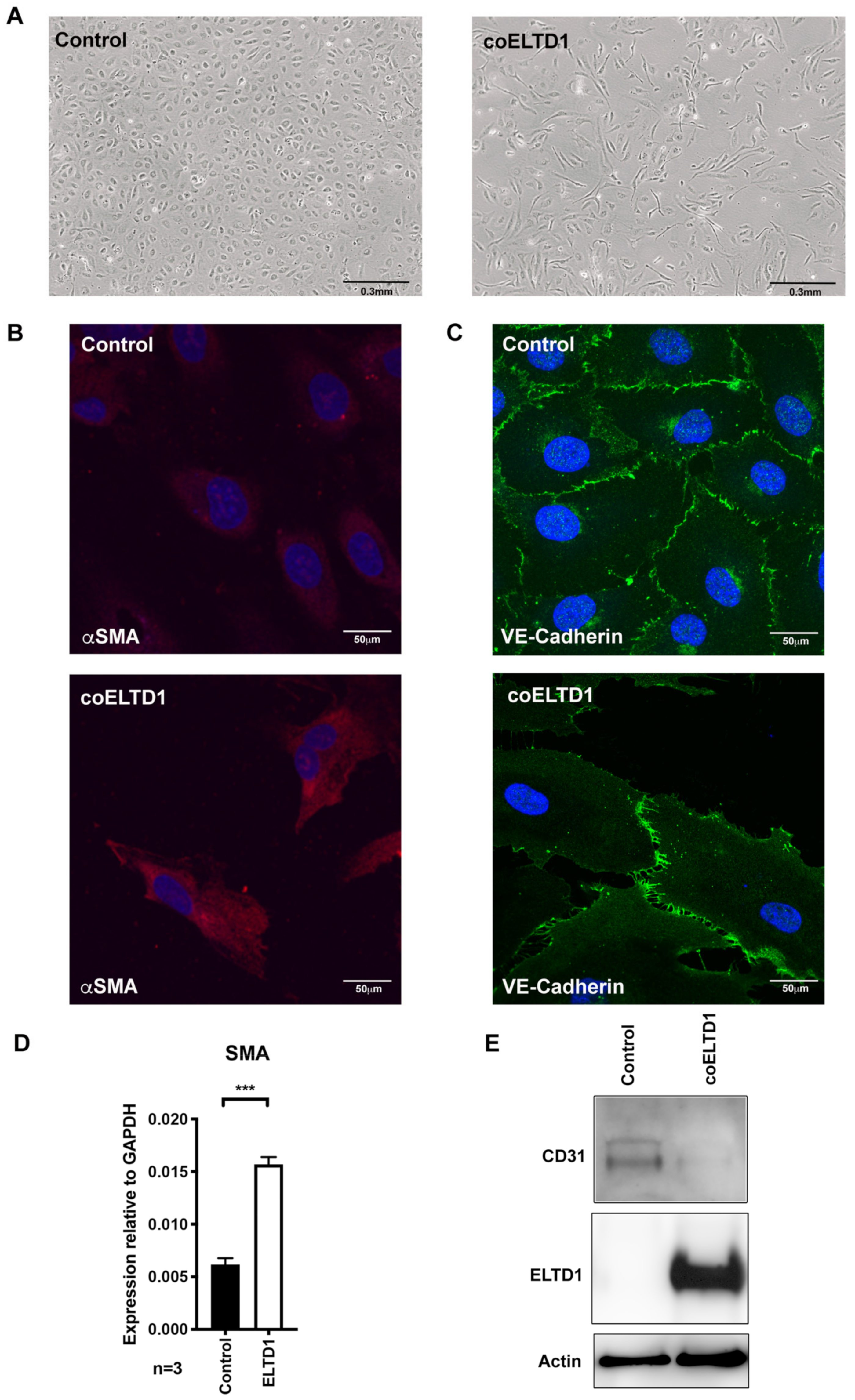
2.1. Expression of coELTD1 in Endothelial Cells
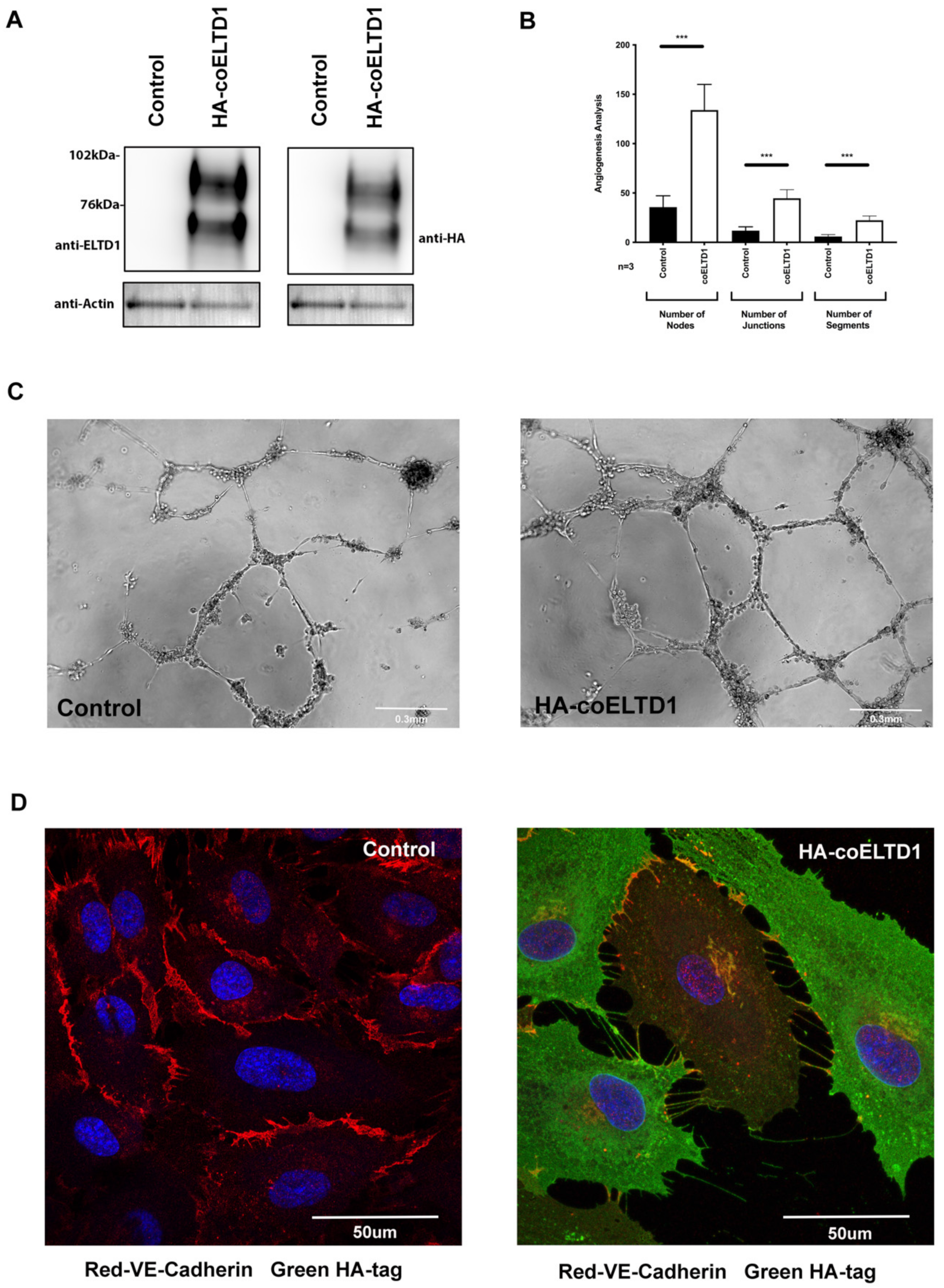
2.2. Expression of Tagged coELTD1
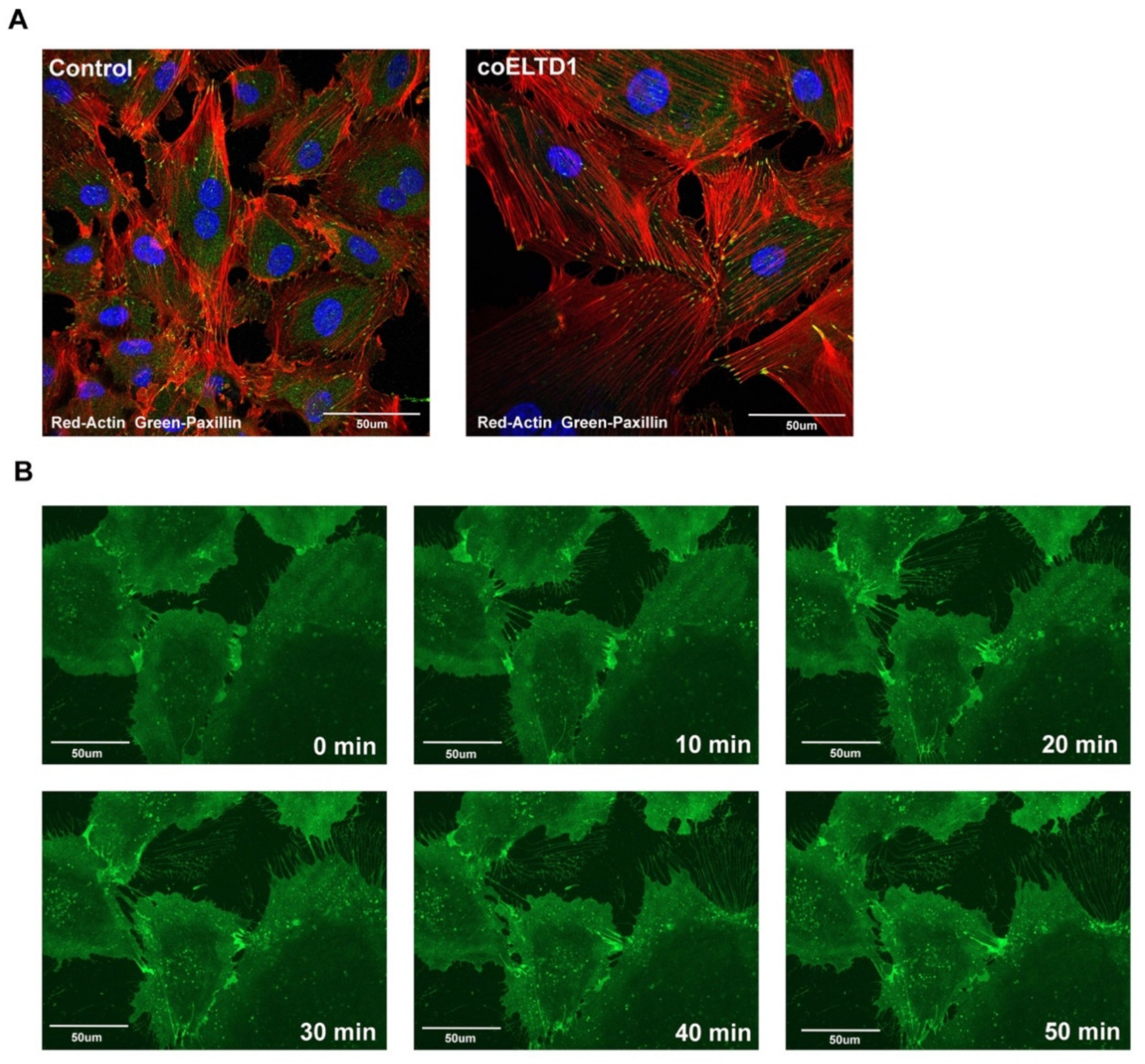
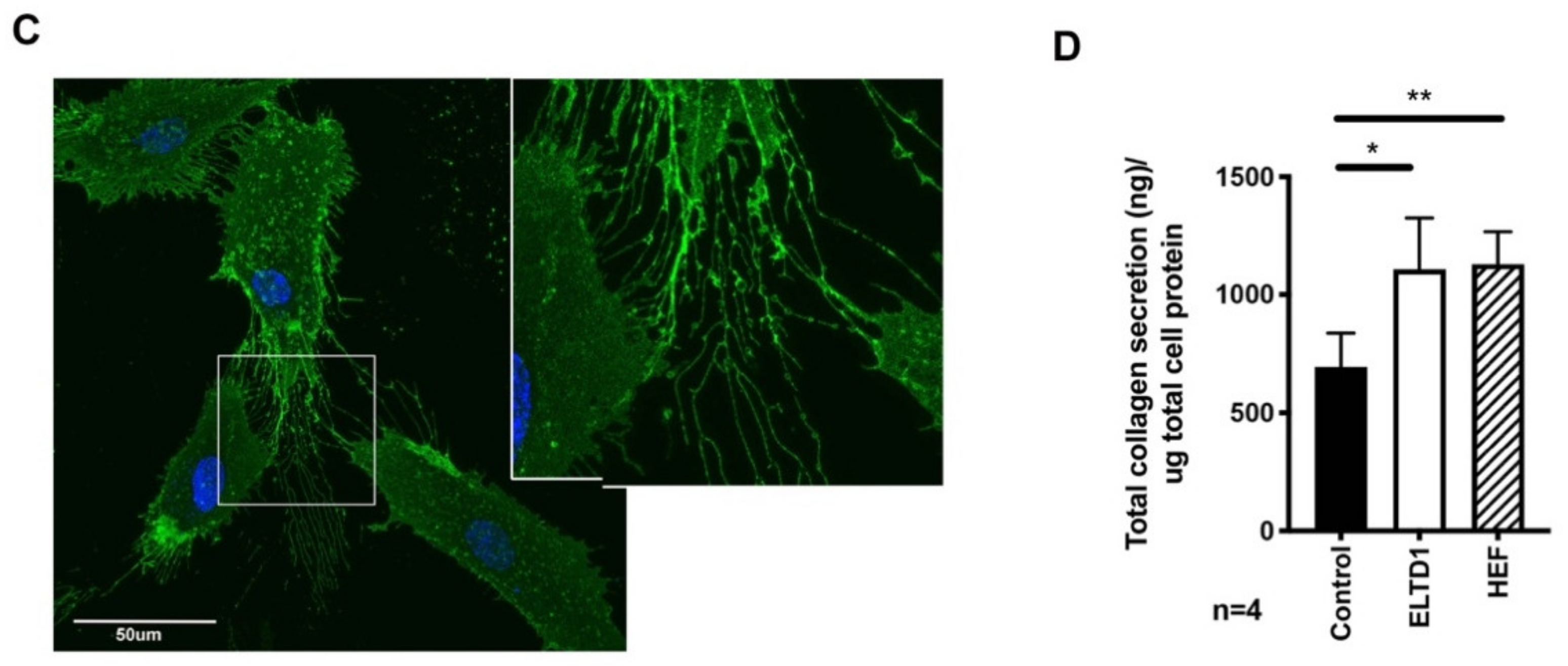
2.3. Confocal Microscopy of coELTD1-Induced Shape Changes
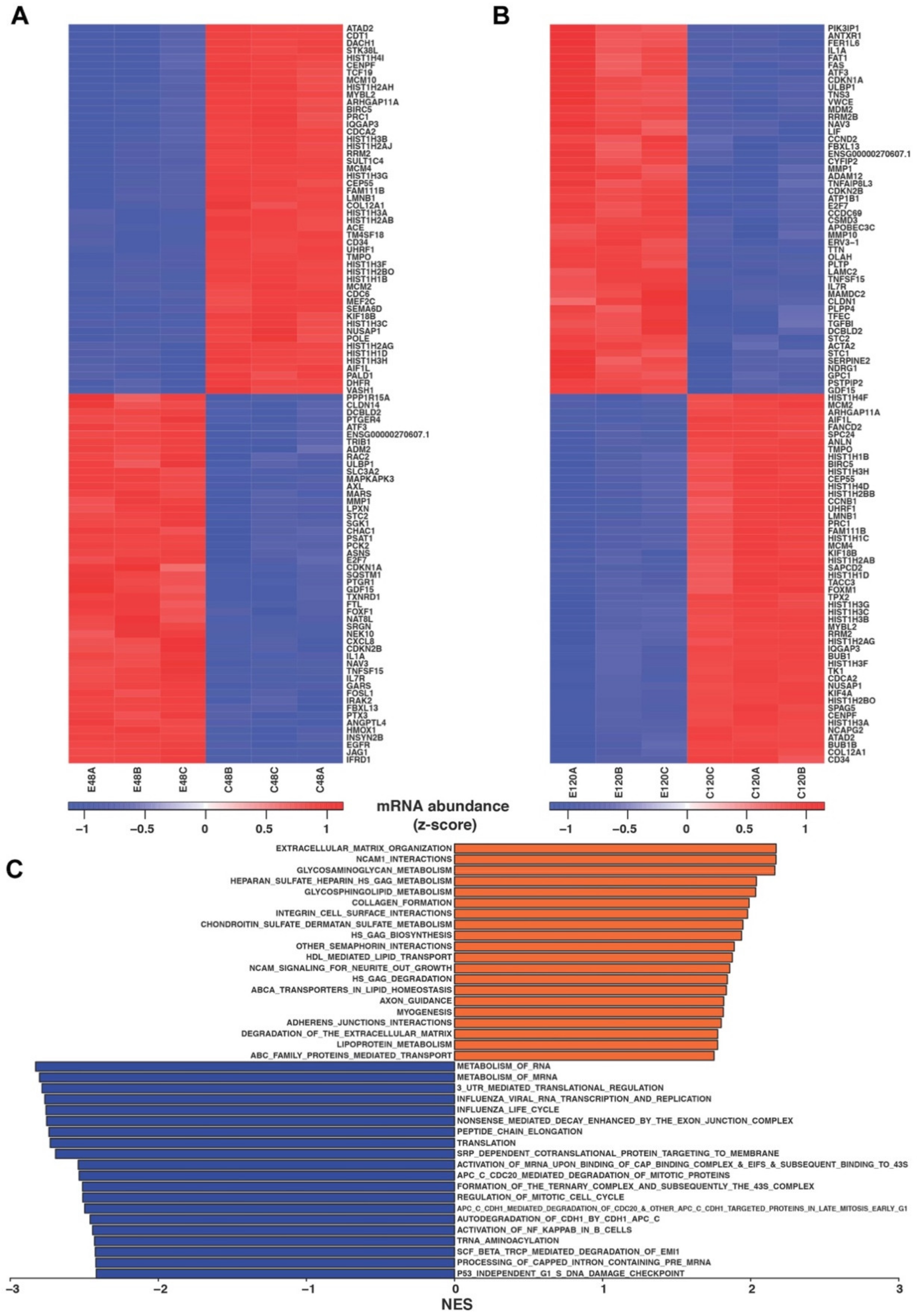
2.4. RNA-Seq of coELTD1 mRNA Expression Profiles
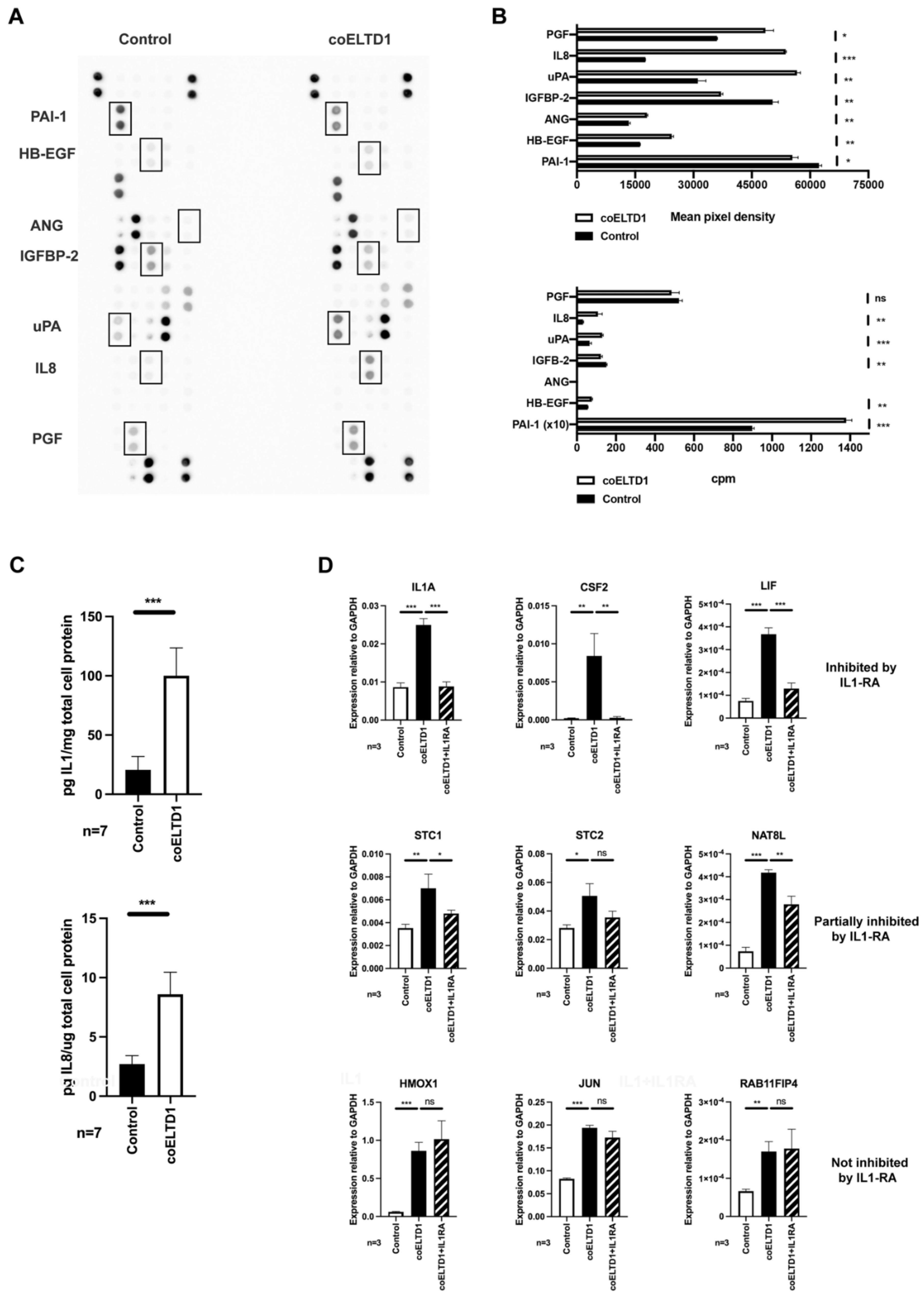
2.5. Factors Effecting Angiogenesis and EndMT to Myofibroblasts
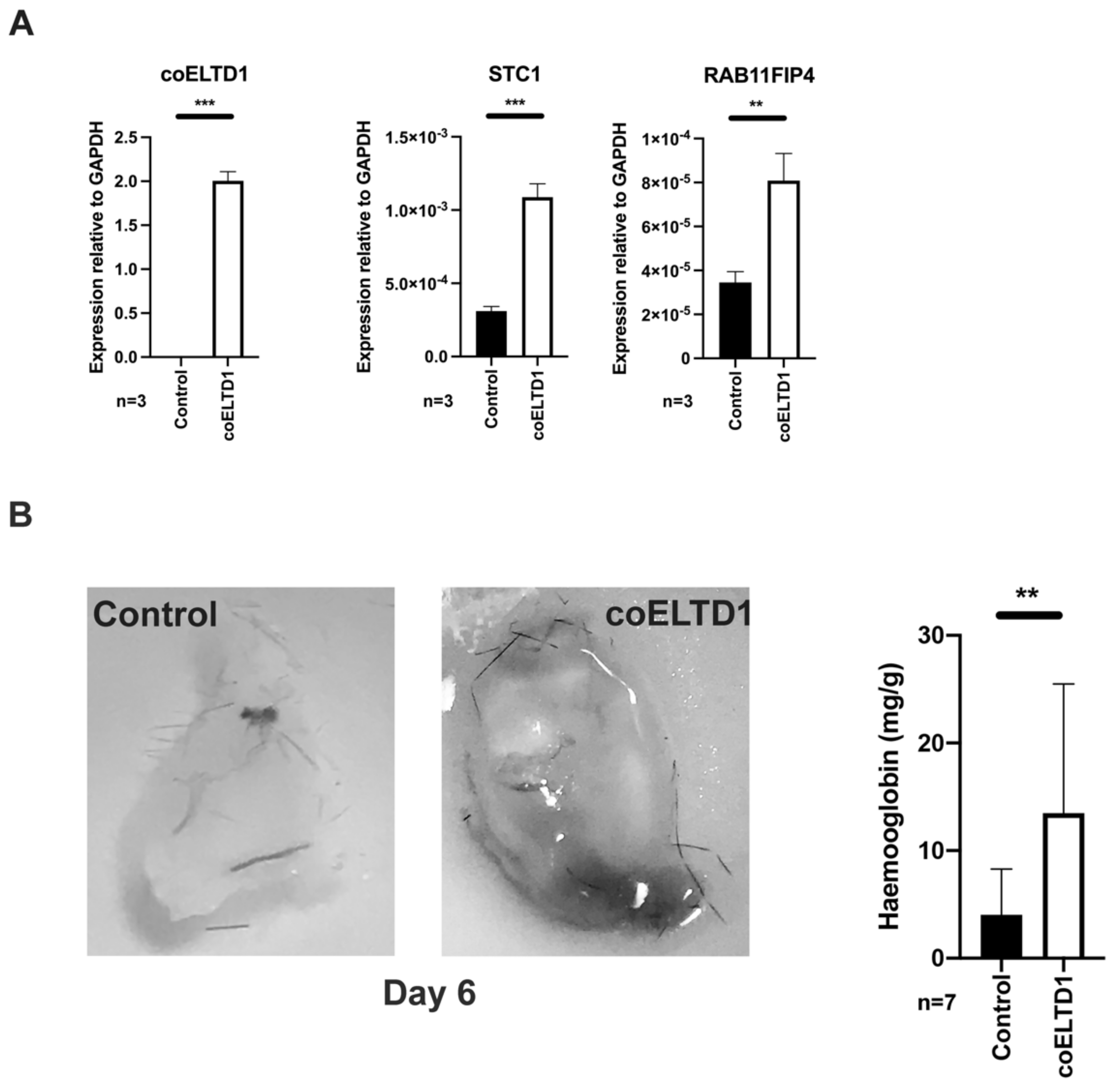
2.6. In Vivo Effects of ELTD1 Expression
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Cell Matrix Adhesion Assay
4.3. Cell-Cell Adhesion Assay
4.4. Migration Assay
4.5. Proliferation Assay
4.6. Hanging Drop Endothelial Sprouting Assay
4.7. Tube Formation Assay
4.8. QPCR Protocol
4.9. Western Blotting
4.10. Immunohistochemistry (IHC) and Immunocytochemistry (ICC)
4.11. Live Cell Imaging
4.12. Collagen Assay
4.13. ELISA
4.14. Angiogenesis Array
4.15. RNA-Seq
4.16. In Vivo MatrigelTM Plug Assay
4.17. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nechiporuk, T.; Urness, L.D.; Keating, M.T. ETL a novel seven-transmembrane receptor that is developmentally regulated in the heart. ETL is a member of the secretin family and belongs to the epidermal growth factor-seven-transmembrane subfamily. J. Biol. Chem. 2001, 276, 4150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yona, S.; Lin, H.H.; Siu, W.O.; Gordon, S.; Stacey, M. Adhesion-GPCRs: Emerging roles for novel receptors. Trends Biochem. Sci. 2008, 33, 491–500. [Google Scholar] [CrossRef]
- Arac, D.; Boucard, A.A.; Bolliger, M.F.; Nguyen, J.; Soltis, S.M.; Sudhof, T.C.; Brunger, A.T. A novel evolutionarily conserved domain of cell-adhesion GPCRs mediates autoproteolysis. EMBO J. 2012, 31, 1364–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cullen, M.; Elzarrad, M.K.; Seaman, S.; Zudaire, E.; Stevens, J.; Yang, M.Y.; Li, X.; Chaudhary, A.; Xu, L.; Hilton, M.B.; et al. GPR124, an orphan G protein-coupled receptor, is required for CNS-specific vascularization and establishment of the blood-brain barrier. Proc. Natl. Acad. Sci. USA 2011, 108, 5759–5764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Ward, Y.; Tian, L.; Lake, R.; Guedez, L.; Stetler-Stevenson, W.G.; Kelly, K. CD97, an adhesion receptor on inflammatory cells, stimulates angiogenesis through binding integrin counterreceptors on endothelial cells. Blood 2005, 105, 2836–2844. [Google Scholar] [CrossRef] [PubMed]
- Niaudet, C.; Hofmann, J.J.; Mae, M.A.; Jung, B.; Gaengel, K.; Vanlandewijck, M.; Ekvarn, E.; Salvado, M.D.; Mehlem, A.; Al Sayegh, S.; et al. Gpr116 Receptor Regulates Distinctive Functions in Pneumocytes and Vascular Endothelium. PLoS ONE 2015, 10, e0137949. [Google Scholar] [CrossRef]
- Wallgard, E.; Larsson, E.; He, L.; Hellstrom, M.; Armulik, A.; Nisancioglu, M.H.; Genove, G.; Lindahl, P.; Betsholtz, C. Identification of a core set of 58 gene transcripts with broad and specific expression in the microvasculature. Arter. Thromb. Vasc. Biol. 2008, 28, 1469–1476. [Google Scholar] [CrossRef] [Green Version]
- Masiero, M.; Simoes, F.C.; Han, H.D.; Snell, C.; Peterkin, T.; Bridges, E.; Mangala, L.S.; Wu, S.Y.; Pradeep, S.; Li, D.; et al. A core human primary tumor angiogenesis signature identifies the endothelial orphan receptor ELTD1 as a key regulator of angiogenesis. Cancer Cell 2013, 24, 229–241. [Google Scholar] [CrossRef] [Green Version]
- Towner, R.A.; Jensen, R.L.; Colman, H.; Vaillant, B.; Smith, N.; Casteel, R.; Saunders, D.; Gillespie, D.L.; Silasi-Mansat, R.; Lupu, F.; et al. ELTD1, a potential new biomarker for gliomas. Neurosurgery 2013, 72, 77–90. [Google Scholar] [CrossRef] [Green Version]
- Ziegler, J.; Pody, R.; Coutinho de Souza, P.; Evans, B.; Saunders, D.; Smith, N.; Mallory, S.; Njoku, C.; Dong, Y.; Chen, H.; et al. ELTD1, an effective anti-angiogenic target for gliomas: Preclinical assessment in mouse GL261 and human G55 xenograft glioma models. Neuro. Oncol. 2017, 19, 175–185. [Google Scholar] [CrossRef]
- Simundza, J.; Cowin, P. Adhesion G-protein-coupled receptors: Elusive hybrids come of age. Cell Commun. Adhes. 2013, 20, 213–226. [Google Scholar] [CrossRef] [Green Version]
- Gupte, J.; Swaminath, G.; Danao, J.; Tian, H.; Li, Y.; Wu, X. Signaling property study of adhesion G-protein-coupled receptors. FEBS Lett. 2012, 586, 1214–1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bond, R.A.; Ijzerman, A.P. Recent developments in constitutive receptor activity and inverse agonism, and their potential for GPCR drug discovery. Trends Pharm. Sci. 2006, 27, 92–96. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van De Water, L.; Varney, S.; Tomasek, J.J. Mechanoregulation of the Myofibroblast in Wound Contraction, Scarring, and Fibrosis: Opportunities for New Therapeutic Intervention. Adv. Wound Care 2013, 2, 122–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirfel, G.; Rigort, A.; Borm, B.; Herzog, V. Cell migration: Mechanisms of rear detachment and the formation of migration tracks. Eur. J. Cell Biol. 2004, 83, 717–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klingberg, F.; Hinz, B.; White, E.S. The myofibroblast matrix: Implications for tissue repair and fibrosis. J. Pathol. 2013, 229, 298–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [Green Version]
- Cho, J.G.; Lee, A.; Chang, W.; Lee, M.S.; Kim, J. Endothelial to Mesenchymal Transition Represents a Key Link in the Interaction between Inflammation and Endothelial Dysfunction. Front. Immunol. 2018, 9, 294. [Google Scholar] [CrossRef] [Green Version]
- Kovacic, J.C.; Mercader, N.; Torres, M.; Boehm, M.; Fuster, V. Epithelial-to-mesenchymal and endothelial-to-mesenchymal transition: From cardiovascular development to disease. Circulation 2012, 125, 1795–1808. [Google Scholar] [CrossRef] [Green Version]
- Kovacic, J.C.; Dimmeler, S.; Harvey, R.P.; Finkel, T.; Aikawa, E.; Krenning, G.; Baker, A.H. Endothelial to Mesenchymal Transition in Cardiovascular Disease: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 73, 190–209. [Google Scholar] [CrossRef]
- Lu, S.; Liu, S.; Wietelmann, A.; Kojonazarov, B.; Atzberger, A.; Tang, C.; Schermuly, R.T.; Grone, H.J.; Offermanns, S. Developmental vascular remodeling defects and postnatal kidney failure in mice lacking Gpr116 (Adgrf5) and Eltd1 (Adgrl4). PLoS ONE 2017, 12, e0183166. [Google Scholar] [CrossRef] [Green Version]
- Gurtner, G.C.; Werner, S.; Barrandon, Y.; Longaker, M.T. Wound repair and regeneration. Nature 2008, 453, 314–321. [Google Scholar] [CrossRef]
- Micallef, L.; Vedrenne, N.; Billet, F.; Coulomb, B.; Darby, I.A.; Desmouliere, A. The myofibroblast, multiple origins for major roles in normal and pathological tissue repair. Fibrogenesis Tissue Repair 2012, 5, S5. [Google Scholar] [CrossRef] [Green Version]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Zalewski, A.; Shi, Y. Vascular myofibroblasts. Lessons from coronary repair and remodeling. Arter. Thromb. Vasc. Biol. 1997, 17, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Jiang, H.; Zhang, R.; Fan, G.; Zhang, Y.; Jiang, D.; Li, H. Augmented cardiac hypertrophy in response to pressure overload in mice lacking ELTD1. PLoS ONE 2012, 7, e35779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goffin, J.M.; Pittet, P.; Csucs, G.; Lussi, J.W.; Meister, J.J.; Hinz, B. Focal adhesion size controls tension-dependent recruitment of alpha-smooth muscle actin to stress fibers. J. Cell Biol. 2006, 172, 259–268. [Google Scholar] [CrossRef] [Green Version]
- Gaggioli, C.; Hooper, S.; Hidalgo-Carcedo, C.; Grosse, R.; Marshall, J.F.; Harrington, K.; Sahai, E. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat. Cell Biol. 2007, 9, 1392–1400. [Google Scholar] [CrossRef]
- Hinz, B.; Celetta, G.; Tomasek, J.J.; Gabbiani, G.; Chaponnier, C. Alpha-smooth muscle actin expression upregulates fibroblast contractile activity. Mol. Biol. Cell 2001, 12, 2730–2741. [Google Scholar] [CrossRef] [Green Version]
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnier, C.; Brown, R.A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2002, 3, 349–363. [Google Scholar] [CrossRef]
- LeWinter, M.M.; Wu, Y.; Labeit, S.; Granzier, H. Cardiac titin: Structure, functions and role in disease. Clin. Chim. Acta 2007, 375, 1–9. [Google Scholar] [CrossRef]
- Cox, T.R.; Erler, J.T. Molecular pathways: Connecting fibrosis and solid tumor metastasis. Clin. Cancer Res. 2014, 20, 3637–3643. [Google Scholar] [CrossRef] [Green Version]
- Zeisberg, E.M.; Potenta, S.; Xie, L.; Zeisberg, M.; Kalluri, R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007, 67, 10123–10128. [Google Scholar] [CrossRef] [Green Version]
- Arciniegas, E.; Frid, M.G.; Douglas, I.S.; Stenmark, K.R. Perspectives on endothelial-to-mesenchymal transition: Potential contribution to vascular remodeling in chronic pulmonary hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 293, L1–L8. [Google Scholar] [CrossRef] [Green Version]
- Teekakirikul, P.; Eminaga, S.; Toka, O.; Alcalai, R.; Wang, L.; Wakimoto, H.; Nayor, M.; Konno, T.; Gorham, J.M.; Wolf, C.M.; et al. Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires Tgf-beta. J. Clin. Investig. 2010, 120, 3520–3529. [Google Scholar] [CrossRef] [Green Version]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [PubMed]
- Sappino, A.P.; Skalli, O.; Jackson, B.; Schurch, W.; Gabbiani, G. Smooth-muscle differentiation in stromal cells of malignant and non-malignant breast tissues. Int. J. Cancer 1988, 41, 707–712. [Google Scholar] [CrossRef] [PubMed]
- De Wever, O.; Demetter, P.; Mareel, M.; Bracke, M. Stromal myofibroblasts are drivers of invasive cancer growth. Int. J. Cancer 2008, 123, 2229–2238. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; Kalluri, R. A peek into cancer-associated fibroblasts: Origins, functions and translational impact. Dis. Model. Mech. 2018, 11, dmm029447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ongusaha, P.P.; Kwak, J.C.; Zwible, A.J.; Macip, S.; Higashiyama, S.; Taniguchi, N.; Fang, L.; Lee, S.W. HB-EGF is a potent inducer of tumor growth and angiogenesis. Cancer Res. 2004, 64, 5283–5290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmeliet, P.; Moons, L.; Luttun, A.; Vincenti, V.; Compernolle, V.; De Mol, M.; Wu, Y.; Bono, F.; Devy, L.; Beck, H.; et al. Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nat. Med. 2001, 7, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Breuss, J.M.; Uhrin, P. VEGF-initiated angiogenesis and the uPA/uPAR system. Cell Adh. Migr. 2012, 6, 535–615. [Google Scholar] [CrossRef] [Green Version]
- Oxvig, C. The role of PAPP-A in the IGF system: Location, location, location. J. Cell Commun. Signal 2015, 9, 177–187. [Google Scholar] [CrossRef] [Green Version]
- Turner, N.A.; Warburton, P.; O’Regan, D.J.; Ball, S.G.; Porter, K.E. Modulatory effect of interleukin-1alpha on expression of structural matrix proteins, MMPs and TIMPs in human cardiac myofibroblasts: Role of p38 MAP kinase. Matrix Biol. 2010, 29, 613–620. [Google Scholar] [CrossRef]
- Russo, R.C.; Garcia, C.C.; Teixeira, M.M.; Amaral, F.A. The CXCL8/IL-8 chemokine family and its receptors in inflammatory diseases. Expert Rev. Clin. Immunol. 2014, 10, 593–619. [Google Scholar] [CrossRef] [Green Version]
- Voronov, E.; Carmi, Y.; Apte, R.N. The role IL-1 in tumor-mediated angiogenesis. Front. Physiol. 2014, 5, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweighofer, B.; Testori, J.; Sturtzel, C.; Sattler, S.; Mayer, H.; Wagner, O.; Bilban, M.; Hofer, E. The VEGF-induced transcriptional response comprises gene clusters at the crossroad of angiogenesis and inflammation. Thromb. Haemost. 2009, 102, 544–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohr, T.; Haudek-Prinz, V.; Slany, A.; Grillari, J.; Micksche, M.; Gerner, C. Proteome profiling in IL-1beta and VEGF-activated human umbilical vein endothelial cells delineates the interlink between inflammation and angiogenesis. PLoS ONE 2017, 12, e0179065. [Google Scholar] [CrossRef] [Green Version]
- Mayer, H.; Bilban, M.; Kurtev, V.; Gruber, F.; Wagner, O.; Binder, B.R.; de Martin, R. Deciphering regulatory patterns of inflammatory gene expression from interleukin-1-stimulated human endothelial cells. Arter. Thromb. Vasc. Biol. 2004, 24, 1192–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keeley, E.C.; Mehrad, B.; Strieter, R.M. Chemokines as mediators of tumor angiogenesis and neovascularization. Exp. Cell Res. 2011, 317, 685–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshiko, Y.; Aubin, J.E. Stanniocalcin 1 as a pleiotropic factor in mammals. Peptides 2004, 25, 1663–1669. [Google Scholar] [CrossRef] [PubMed]
- Ohkouchi, S.; Ono, M.; Kobayashi, M.; Hirano, T.; Tojo, Y.; Hisata, S.; Ichinose, M.; Irokawa, T.; Ogawa, H.; Kurosawa, H. Myriad Functions of Stanniocalcin-1 (STC1) Cover Multiple Therapeutic Targets in the Complicated Pathogenesis of Idiopathic Pulmonary Fibrosis (IPF). Clin. Med. Insights Circ. Respir Pulm. Med. 2015, 9, 91–96. [Google Scholar] [CrossRef]
- Chen, F.; Zhang, Z.; Pu, F. Role of stanniocalcin-1 in breast cancer. Oncol. Lett. 2019, 18, 3946–3953. [Google Scholar]
- Pessentheiner, A.R.; Pelzmann, H.J.; Walenta, E.; Schweiger, M.; Groschner, L.N.; Graier, W.F.; Kolb, D.; Uno, K.; Miyazaki, T.; Nitta, A.; et al. NAT8L (N-acetyltransferase 8-like) accelerates lipid turnover and increases energy expenditure in brown adipocytes. J. Biol. Chem. 2013, 288, 36040–36051. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.K.; Johnson, L.C.; Clodfelter, J.E.; Pemble, C.W.T.; Fulp, B.E.; Furdui, C.M.; Kridel, S.J.; Lowther, W.T. Crystal Structure and Substrate Specificity of Human Thioesterase 2: Insights Into the Molecular Basis for the Modulation of Fatty Acid Synthese. J. Biol. Chem. 2016, 291, 3520–3530. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Yazdani, S.; Bansal, R.; Prakash, J. Drug targeting to myofibroblasts: Implications for fibrosis and cancer. Adv. Drug Deliv. Rev. 2017, 121, 101–116. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Oon, C.E.; Bridges, E.; Sheldon, H.; Sainson, R.C.A.; Jubb, A.; Turley, H.; Leek, R.; Buffa, F.; Harris, A.L.; Li, J.L. Role of Delta-like 4 in Jagged1-induced tumour angiogenesis and tumour growth. Oncotarget 2017, 8, 40115–40131. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, S.; Li, W. RSeQC: Quality control of RNA-seq experiments. Bioinformatics 2012, 28, 2184–2185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sheldon, H.; Alexander, J.; Bridges, E.; Moreira, L.; Reilly, S.; Ang, K.H.; Wang, D.; Lin, S.; Haider, S.; Banham, A.H.; et al. ELTD1 Activation Induces an Endothelial-EMT Transition to a Myofibroblast Phenotype. Int. J. Mol. Sci. 2021, 22, 11293. https://doi.org/10.3390/ijms222011293
Sheldon H, Alexander J, Bridges E, Moreira L, Reilly S, Ang KH, Wang D, Lin S, Haider S, Banham AH, et al. ELTD1 Activation Induces an Endothelial-EMT Transition to a Myofibroblast Phenotype. International Journal of Molecular Sciences. 2021; 22(20):11293. https://doi.org/10.3390/ijms222011293
Chicago/Turabian StyleSheldon, Helen, John Alexander, Esther Bridges, Lucia Moreira, Svetlana Reilly, Koon Hwee Ang, Dian Wang, Salwa Lin, Syed Haider, Alison H. Banham, and et al. 2021. "ELTD1 Activation Induces an Endothelial-EMT Transition to a Myofibroblast Phenotype" International Journal of Molecular Sciences 22, no. 20: 11293. https://doi.org/10.3390/ijms222011293