
Aberrant Mitochondrial Dynamics and Exacerbated Response to Neuroinflammation in a Novel Mouse Model of CMT2A

 , , , ,
, , , ,  , ,
, , 
Abstract
:1. Introduction
2. Results
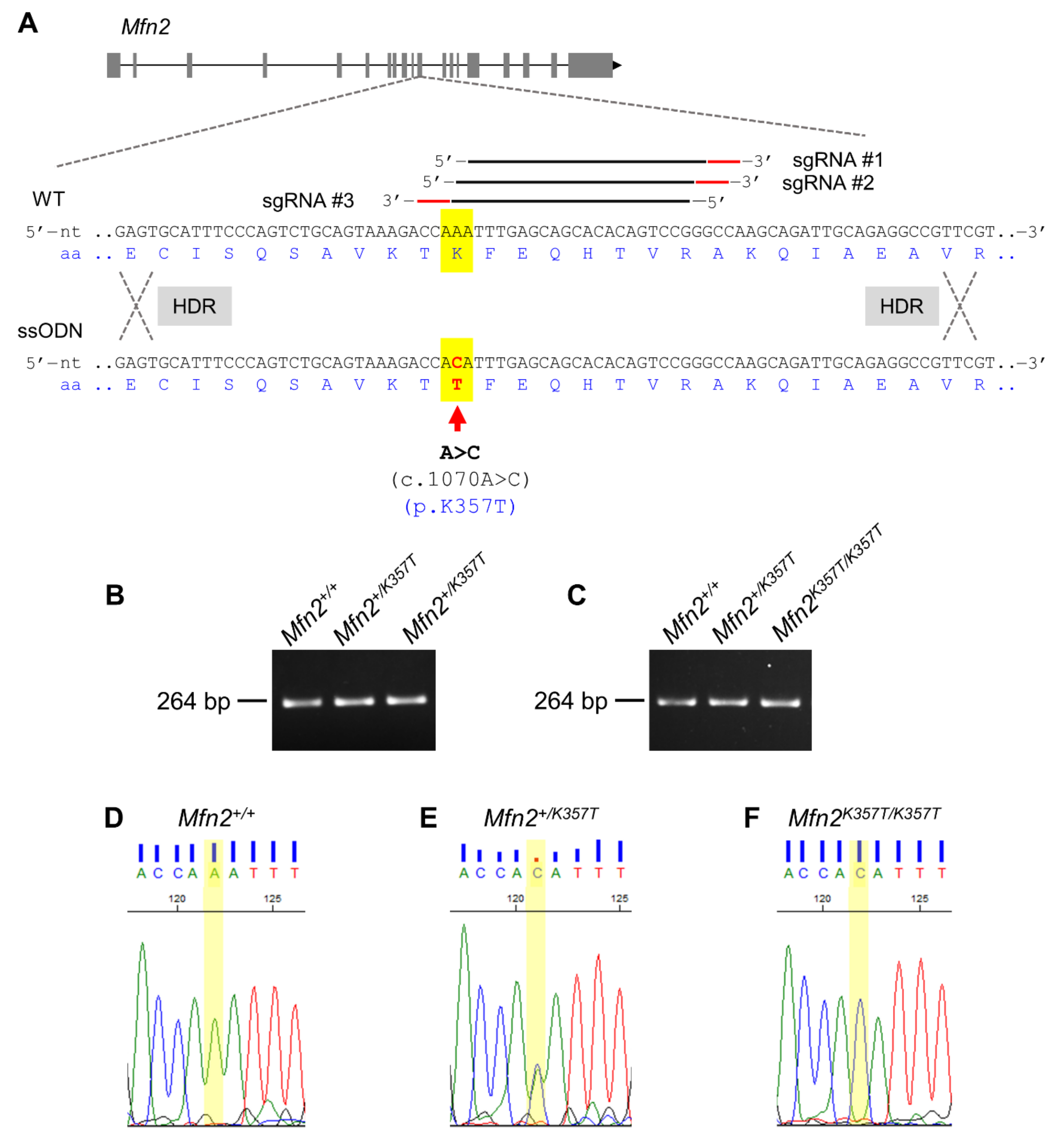
2.1. Replication of the Human MFN2K357T Mutation in a Mfn2K357T-Expressing CMT2A KI Mouse Model
2.2. Mfn2K357T/K357T Homozygous Mouse Pups Are Susceptible to Postnatal Lethality and Present with Severe Mitochondrial Clustering
2.3. Behavioral Performance and Sciatic Nerve Electrophysiological Properties of Mfn2+/K357T Mice Are Not Affected up to 10 mo of Age
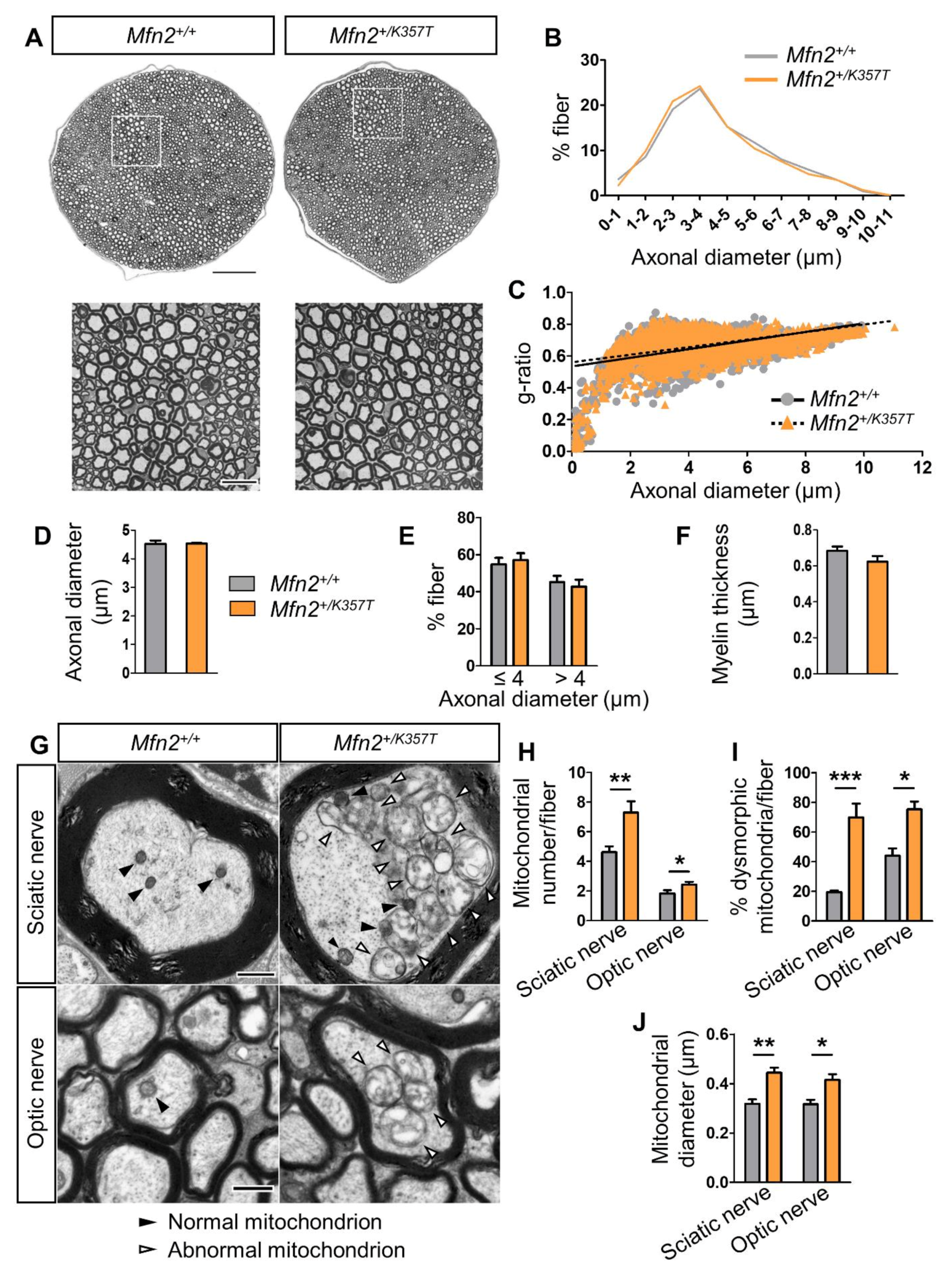
2.4. Histopathological Changes in Sciatic and Optic Nerves of Mfn2+/K357T Mice
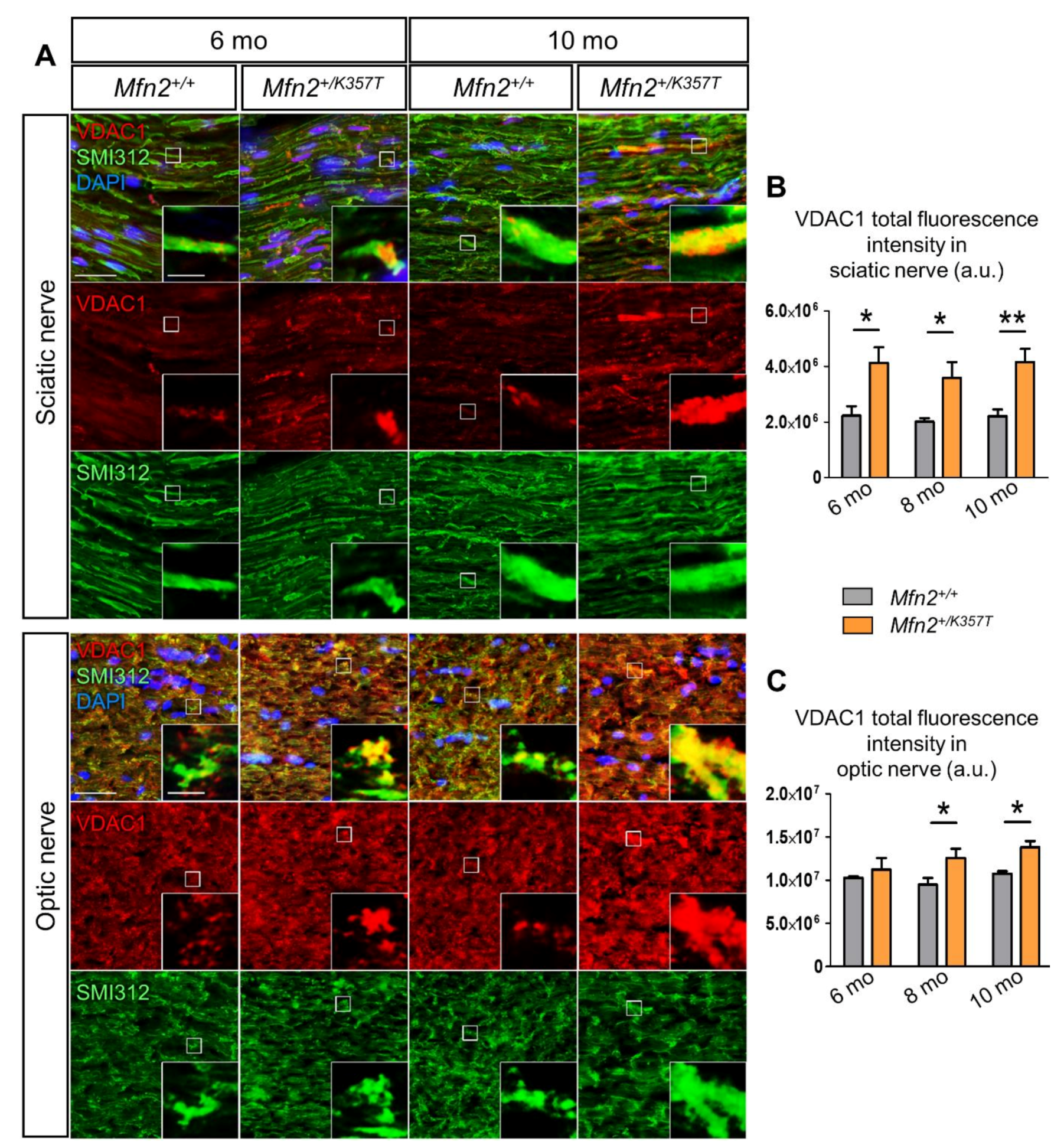
2.5. Mitochondrial Clustering Increases with Age and Is More Pronounced in Sciatic and Optic Nerves of Mfn2+/K357T Mice
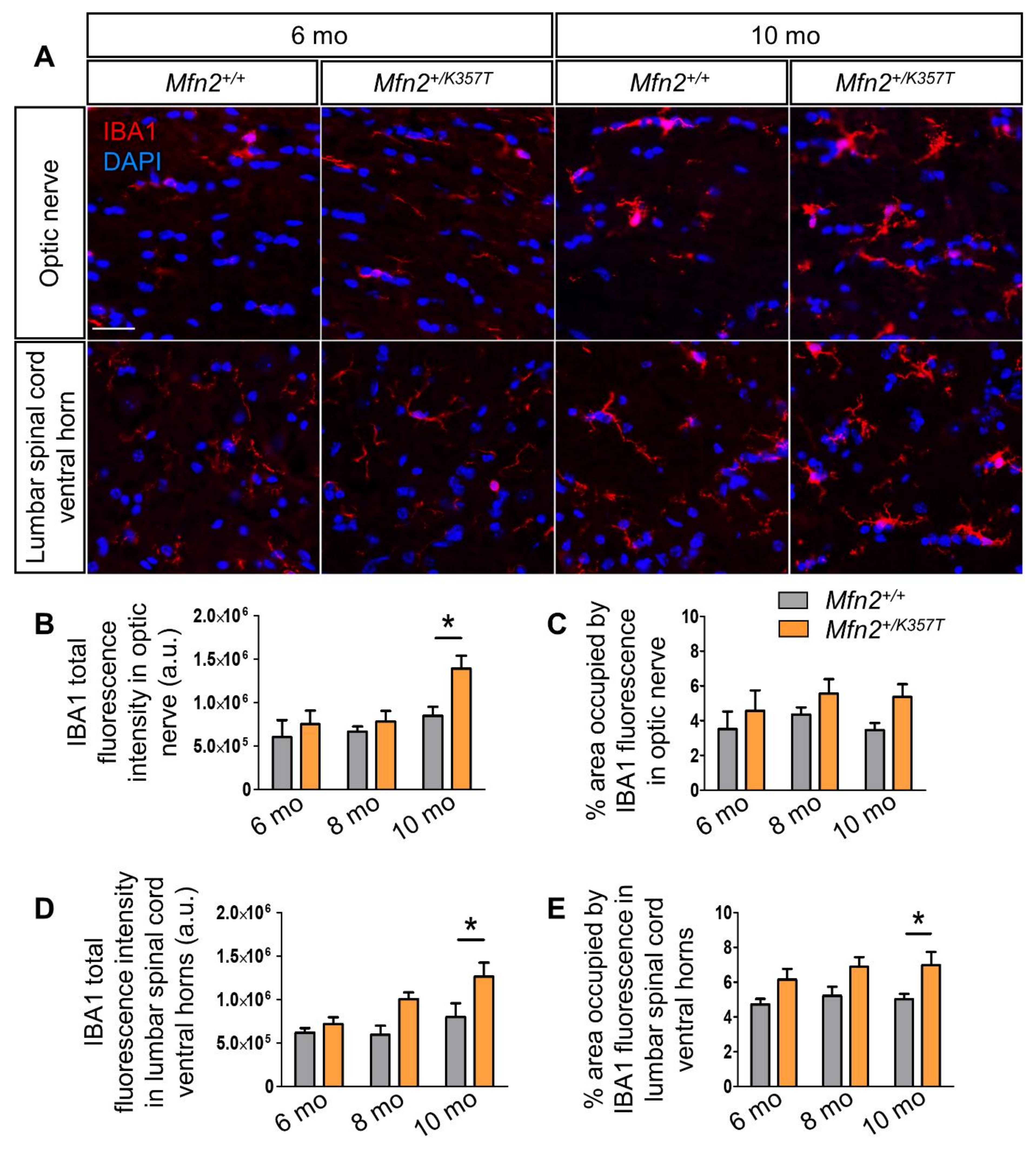
2.6. Microgliosis but Not Astrogliosis in the CNS of 10-mo-old Mfn2+/K357T Mice
2.7. Impaired Behavioral Performance and Increased Peripheral Inflammatory Response in Mfn2+/K357T Mice Compared to Mfn2+/+ Mice at 4 h Post-LPS
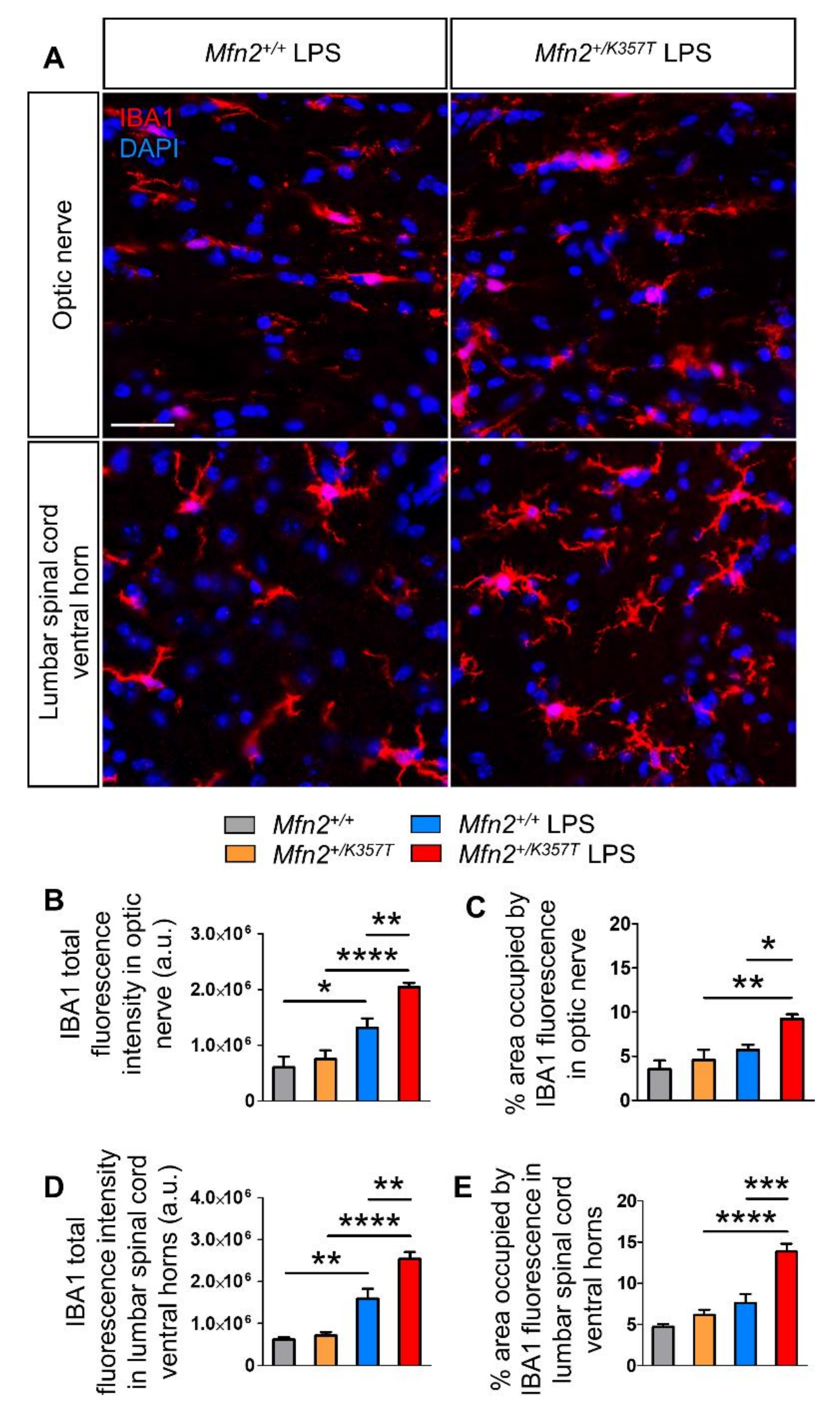
2.8. Pronounced Microgliosis in the CNS of Mfn2+/K357T Mice Compared to Mfn2+/+ Mice at 96 h Post-LPS
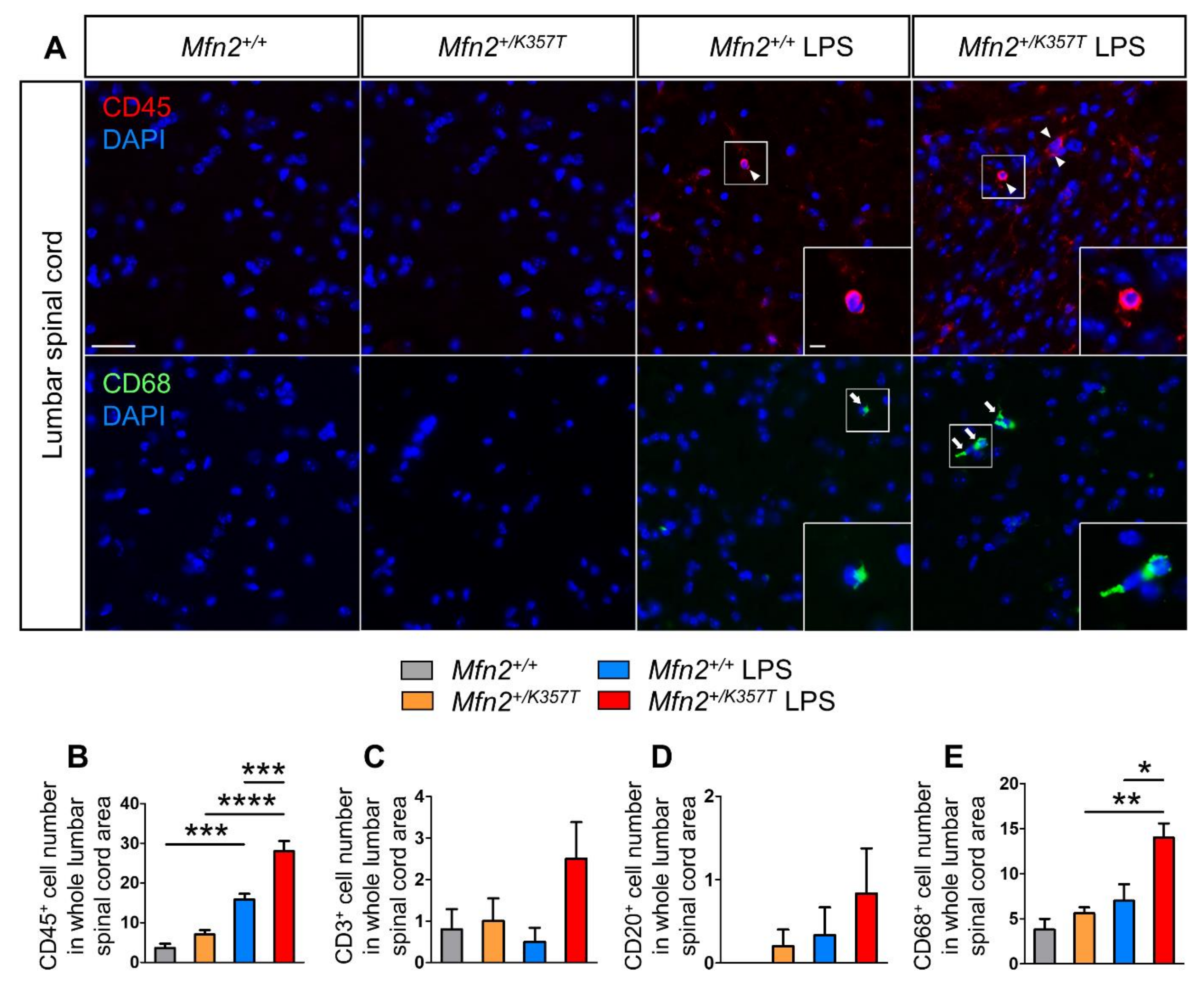
2.9. Increased Infiltration of Peripheral Leukocytes, Especially Macrophages, into the CNS of Mfn2+/K357T Compared to Mfn2+/+ Mice 96 h Post-LPS
2.10. No Significant Alterations in Mitochondrial Dynamics in Mfn2+/+ and Mfn2+/K357T Mice 96 h after LPS Injection
3. Discussion
4. Materials and Methods
4.1. Generation of CMT2A KI Mice Expressing Mfn2K357T
4.2. Behavioral Testing
4.3. Sciatic Nerve Motor Conduction Velocity Studies
4.4. LPS-Induced Systemic Inflammation
4.5. Blood Collection and Sandwich ELISA Analysis of Serum Cytokines
4.6. Tissue Processing and Immunofluorescence Staining
4.7. Tissue Processing and Morphometric Analyses of Sciatic Nerves
4.8. Transmission Electron Microscopy Study of Sciatic and Optic Nerve Mitochondria
4.9. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Skre, H. Genetic and clinical aspects of Charcot-Marie-Tooth’s disease. Clin. Genet. 1974, 6, 98–118. [Google Scholar] [CrossRef] [PubMed]
- Kurihara, S.; Adachi, Y.; Wada, K.; Awaki, E.; Harada, H.; Nakashima, K. An epidemiological genetic study of Charcot-Marie-Tooth disease in Western Japan. Neuroepidemiology 2002, 21, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Foley, C.; Schofield, I.; Eglon, G.; Bailey, G.; Chinnery, P.F.; Horvath, R. Charcot-Marie-Tooth disease in North-ern England. J. Neurol. Neurosurg Psychiatry 2012, 83, 572–573. [Google Scholar] [CrossRef] [PubMed]
- Theadom, A.; Roxburgh, R.; MacAulay, E.; O’Grady, G.; Burns, J.; Parmar, P.; Jones, K.; Rodrigues, M. Prevalence of Charcot-Marie-Tooth disease across the lifespan: A population-based epidemiological study. BMJ Open 2019, 9, e029240. [Google Scholar] [CrossRef]
- Ando, M.; Hashiguchi, A.; Okamoto, Y.; Yoshimura, A.; Hiramatsu, Y.; Yuan, J.; Higuchi, Y.; Mitsui, J.; Ishiura, H.; Umemura, A.; et al. Clinical and genetic diversities of Charcot-Marie-Tooth disease with MFN2 mutations in a large case study. J. Peripher. Nerv. Syst. 2017, 22, 191–199. [Google Scholar] [CrossRef] [Green Version]
- Chung, K.W.; Kim, S.B.; Park, K.D.; Choi, K.G.; Lee, J.H.; Eun, H.W.; Suh, J.S.; Hwang, J.H.; Kim, W.K.; Seo, B.C.; et al. Early onset severe and late-onset mild Charcot-Marie-Tooth disease with mitofusin 2 (MFN2) mutations. Brain 2006, 129, 2103–2118. [Google Scholar] [CrossRef]
- Del Bo, R.; Moggio, M.; Rango, M.; Bonato, S.; D’angelo, M.G.; Ghezzi, S.; Airoldi, G.; Bassi, M.T.; Guglieri, M.; Napoli, L.; et al. Mutated mitofusin 2 presents with intrafamilial variability and brain mitochondrial dysfunction. Neurology 2008, 71, 1959–1966. [Google Scholar]
- Hsu, Y.H.; Lin, K.P.; Guo, Y.C.; Tsai, Y.S.; Liao, Y.C.; Lee, Y.C. Mutation spectrum of Charcot-Marie-Tooth dis-ease among the Han Chinese in Taiwan. Ann. Clin. Transl. Neurol. 2019, 6, 1090–1101. [Google Scholar] [CrossRef] [Green Version]
- Züchner, S.; Mersiyanova, I.V.; Muglia, M.; Bissar-Tadmouri, N.; Rochelle, J.; Dadali, E.L.; Zappia, M.; Nelis, E.; Patitucci, A.; Senderek, J.; et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 2004, 36, 449–451. [Google Scholar] [CrossRef]
- Choi, B.O.; Nakhro, K.; Park, H.J.; Hyun, Y.S.; Lee, J.H.; Kanwal, S.; Jung, S.C.; Chung, K.W. Chung A cohort study of MFN2 mutations and phenotypic spectrums in Charcot-Marie-Tooth disease 2A patients. Clin. Genet. 2015, 87, 594–598. [Google Scholar] [CrossRef]
- Rouzier, C.; Bannwarth, S.; Chaussenot, A.; Chevrollier, A.; Verschueren, A.; Bonello-Palot, N.; Fragaki, K.; Cano, A.; Pouget, J.; Pellissier, J.F.; et al. The MFN2 gene is responsible for mitochondrial DNA instability and optic atrophy ‘plus’ phenotype. Brain 2012, 135, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Rocha, N.; Bulger, D.A.; Frontini, A.; Titheradge, H.; Gribsholt, S.B.; Knox, R.; Page, M.; Harris, J.; Payne, F.; Adams, C.; et al. Human biallelic MFN2 mutations induce mitochondrial dysfunction, upper body adipose hyperplasia, and suppression of leptin expression. Elife 2017, 6, e23813. [Google Scholar] [CrossRef]
- Sawyer, S.L.; Cheuk-Him Ng, A.; Innes, A.M.; Wagner, J.D.; Dyment, D.A.; Tetreault, M.; Care4Rare Canada Consortium; Majewski, J.; Boycott, K.M.; Screaton, R.A.; et al. Homozygous mutations in MFN2 cause multiple symmetric li-pomatosis associated with neuropathy. Hum. Mol. Genet. 2015, 24, 5109–5114. [Google Scholar] [CrossRef] [Green Version]
- Tufano, M.; Cappuccio, G.; Terrone, G.; Manganelli, F.; Pisciotta, C.; Geroldi, A.; Capponi, S.; Del Giudice, E. Early onset Charcot-Marie-Tooth neuropathy type 2A and severe developmental delay: Expanding the clinical phenotype of MFN2-related neuropathy. J. Peripher. Nerv. Syst. 2015, 20, 415–418. [Google Scholar] [CrossRef] [Green Version]
- Santel, A.; Frank, S.; Gaume, B.; Herrler, M.; Youle, R.J.; Fuller, M.T. Mitofusin-1 protein is a generally ex-pressed mediator of mitochondrial fusion in mammalian cells. J. Cell Sci. 2003, 116, 2763–2774. [Google Scholar] [CrossRef] [Green Version]
- Rojo, M.; Legros, F.; Chateau, D.; Lombès, A. Lombes Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mammalian homologs of the transmembrane GTPase Fzo. J. Cell Sci. 2002, 115, 1663–1674. [Google Scholar] [CrossRef]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordi-nately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef]
- Xiong, W.; Ma, Z.; An, D.; Liu, Z.; Cai, W.; Bai, Y.; Zhan, Q.; Lai, W.; Zeng, Q.; Ren, H.; et al. Mitofusin 2 Participates in Mitophagy and Mitochondrial Fusion Against Angiotensin II-Induced Cardiomyocyte Injury. Front. Physiol. 2019, 10, 411. [Google Scholar] [CrossRef]
- De Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef]
- Bernard-Marissal, N.; van Hameren, G.; Juneja, M.; Pellegrino, C.; Louhivuori, L.; Bartesaghi, L.; Rochat, C.; El Mansour, O.; Médard, J.J.; Croisier, M.; et al. Altered interplay between endoplasmic reticulum and mitochondria in Charcot-Marie-Tooth type 2A neuropathy. Proc. Natl. Acad. Sci. USA 2019, 116, 2328–2337. [Google Scholar] [CrossRef] [Green Version]
- Misko, A.; Jiang, S.; Wegorzewska, I.; Milbrandt, J.; Baloh, R.H. Mitofusin 2 is necessary for transport of ax-onal mitochondria and interacts with the Miro/Milton complex. J. Neurosci. 2010, 30, 4232–4240. [Google Scholar] [CrossRef] [Green Version]
- Franco, A.; Kitsis, R.N.; Fleischer, J.A.; Gavathiotis, E.; Kornfeld, O.S.; Gong, G.; Biris, N.; Benz, A.; Qvit, N.; Donnelly, S.K.; et al. Correcting mitochondrial fusion by manipulating mitofusin conformations. Nature 2016, 540, 74–79. [Google Scholar] [CrossRef]
- Honda, S.; Aihara, T.; Hontani, M.; Okubo, K.; Hirose, S. Mutational analysis of action of mitochondrial fusion factor mitofusin-2. J. Cell Sci. 2005, 118, 3153–3161. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.J.; Cao, Y.L.; Feng, J.X.; Qi, Y.; Meng, S.; Yang, J.F.; Zhong, Y.T.; Kang, S.; Chen, X.; Lan, L.; et al. Structural in-sights of human mitofusin-2 into mitochondrial fusion and CMT2A onset. Nat. Commun. 2019, 10, 4914. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.; Qi, Y.; Huang, X.; Yu, C.; Lan, L.; Guo, X.; Rao, Z.; Hu, J.; Lou, Z. Structural basis for GTP hydrolysis and conformational change of MFN1 in mediating membrane fusion. Nat. Struct. Mol. Biol. 2018, 25, 233–243. [Google Scholar] [CrossRef]
- Harland, M.; Torres, S.; Liu, J.; Wang, X. Neuronal Mitochondria Modulation of LPS-Induced Neuroinflamma-tion. J. Neurosci. 2020, 40, 1756–1765. [Google Scholar] [CrossRef]
- Detmer, S.A.; Chan, D.C. Complementation between mouse Mfn1 and Mfn2 protects mitochondrial fusion defects caused by CMT2A disease mutations. J. Cell Biol. 2007, 176, 405–414. [Google Scholar] [CrossRef] [Green Version]
- Bannerman, P.; Burns, T.; Xu, J.; Miers, L.; Pleasure, D. Mice Hemizygous for a Pathogenic Mitofusin-2 Allele Exhibit Hind Limb/Foot Gait Deficits and Phenotypic Perturbations in Nerve and Muscle. PLoS ONE 2016, 11, e0167573. [Google Scholar] [CrossRef]
- Zhou, Y.; Carmona, S.; Muhammad, A.K.M.G.; Bell, S.; Landeros, J.; Vazquez, M.; Ho, R.; Franco, A.; Lu, B.; Dorn, G.W.; et al. Restoring mitofusin balance prevents axonal degeneration in a Charcot-Marie-Tooth type 2A model. J. Clin. Investig. 2019, 130, 1756–1771. [Google Scholar] [CrossRef]
- Wolf, C.; Zimmermann, R.; Thaher, O.; Bueno, D.; Wüllner, V.; Schäfer, M.K.; Albrecht, P.; Methner, A. The Charcot-Marie Tooth Disease Mutation R94Q in MFN2 Decreases ATP Production but Increases Mitochondrial Respiration under Conditions of Mild Oxidative Stress. Cells 2019, 8, 1289. [Google Scholar] [CrossRef] [Green Version]
- Cai, K.C.; van Mil, S.; Murray, E.; Mallet, J.F.; Matar, C.; Ismail, N. Age and sex differences in immune response following LPS treatment in mice. Brain. Behav. Immun. 2016, 58, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Kuo, S.M. Gender Difference in Bacteria Endotoxin-Induced Inflammatory and Anorexic Responses. PLoS ONE 2016, 11, e0162971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosyreva, A.M.; Dzhalilova, D.S.; Makarova, O.V.; Tsvetkov, I.S.; Zolotova, N.A.; Diatroptova, M.A.; Ponomarenko, E.A.; Mkhitarov, V.A.; Khochanskiy, D.N.; Mikhailova, L.P. Sex differences of inflammatory and immune re-sponse in pups of Wistar rats with SIRS. Sci. Rep. 2020, 10, 15884. [Google Scholar] [CrossRef] [PubMed]
- Noh, H.; Jeon, J.; Seo, H. Systemic injection of LPS induces region-specific neuroinflammation and mito-chondrial dysfunction in normal mouse brain. Neurochem. Int. 2014, 69, 35–40. [Google Scholar] [CrossRef]
- Haruwaka, K.; Ikegami, A.; Tachibana, Y.; Ohno, N.; Konishi, H.; Hashimoto, A.; Matsumoto, M.; Kato, D.; Ono, R.; Kiyama, H.; et al. Dual microglia effects on blood brain barrier permeability induced by systemic inflammation. Nat. Commun. 2019, 10, 5816. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Bi, W.; Xiao, S.; Lan, X.; Cheng, X.; Zhang, J.; Lu, D.; Wei, W.; Wang, Y.; Li, H.; et al. Neuroinflammation in-duced by lipopolysaccharide causes cognitive impairment in mice. Sci. Rep. 2019, 9, 5790. [Google Scholar] [CrossRef] [Green Version]
- Rodgers, K.R.; Lin, Y.; Langan, T.J.; Iwakura, Y.; Chou, R.C. Innate Immune Functions of Astrocytes are De-pendent Upon Tumor Necrosis Factor-Alpha. Sci. Rep. 2020, 10, 7047. [Google Scholar] [CrossRef]
- Kang, J.B.; Park, D.J.; Shah, M.A.; Kim, M.O.; Koh, P.O. Lipopolysaccharide induces neuroglia activation and NF-kappaB activation in cerebral cortex of adult mice. Lab. Anim. Res. 2019, 35, 19. [Google Scholar] [CrossRef]
- Becher, B.; Antel, J.P. Comparison of phenotypic and functional properties of immediately ex vivo and cul-tured human adult microglia. Glia 1996, 18, 1–10. [Google Scholar] [CrossRef]
- Fiala, M.; Liu, Q.N.; Sayre, J.; Pop, V.; Brahmandam, V.; Graves, M.C.; Vinters, H.V. Cyclooxygenase-2-positive macrophages infiltrate the Alzheimer’s disease brain and damage the blood-brain barrier. Eur. J. Clin. Invest. 2002, 32, 360–371. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Choi, H.; Min, J.S.; Park, S.J.; Kim, J.H.; Park, H.J.; Kim, B.; Chae, J.I.; Yim, M.; Lee, D.S. Mitochondrial dynamics modulate the expression of pro-inflammatory mediators in microglial cells. J. Neurochem. 2013, 127, 221–232. [Google Scholar] [CrossRef]
- Biel, T.G.; Lee, S.; Flores-Toro, J.A.; Dean, J.W.; Go, K.L.; Lee, M.H.; Law, B.K.; Law, M.E.; Dunn, W.A.; Zendejas, I.; et al. Sirtuin 1 suppresses mitochondrial dysfunction of ischemic mouse livers in a mitofusin 2-dependent man-ner. Cell Death Differ. 2016, 23, 279–290. [Google Scholar] [CrossRef] [Green Version]
- Evans, C.S.; Holzbaur, E.L. Degradation of engulfed mitochondria is rate-limiting in Optineurin-mediated mi-tophagy in neurons. Elife 2020, 9, e50260. [Google Scholar] [CrossRef]
- Cai, Q.; Zakaria, H.M.; Simone, A.; Sheng, Z.H. Spatial parkin translocation and degradation of damaged mi-tochondria via mitophagy in live cortical neurons. Curr. Biol. 2012, 22, 545–552. [Google Scholar] [CrossRef] [Green Version]
- Tur, J.; Pereira-Lopes, S.; Vico, T.; Marín, E.A.; Muñoz, J.P.; Hernández-Alvarez, M.; Cardona, P.J.; Zorzano, A.; Lloberas, J.; Celada, A. Mitofusin 2 in Macrophages Links Mitochondrial ROS Production, Cytokine Release, Phagocytosis, Autophagy, and Bactericidal Activity. Cell Rep. 2020, 32, 108079. [Google Scholar] [CrossRef]
- Riedemann, N.C.; Guo, R.F.; Hollmann, T.J.; Gao, H.; Neff, T.A.; Reuben, J.S.; Speyer, C.L.; Sarma, J.V.; Wetsel, R.A.; Zetoune, F.S.; et al. Regulatory role of C5a in LPS-induced IL-6 production by neutrophils during sepsis. FASEB J. 2004, 18, 370–372. [Google Scholar] [CrossRef]
- Frost, R.A.; Nystrom, G.J.; Lang, C.H. Lipopolysaccharide regulates proinflammatory cytokine expression in mouse myoblasts and skeletal muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002, 283, R698–R709. [Google Scholar] [CrossRef]
- Sundararaj, K.P.; Samuvel, D.J.; Li, Y.; Sanders, J.J.; Lopes-Virella, M.F.; Huang, Y. Interleukin-6 released from fibroblasts is essential for up-regulation of matrix metalloproteinase-1 expression by U937 macrophages in coculture: Cross-talking between fibroblasts and U937 macrophages exposed to high glucose. J. Biol. Chem. 2009, 284, 13714–13724. [Google Scholar] [CrossRef] [Green Version]
- Audoy-Rémus, J.; Richard, J.F.; Soulet, D.; Zhou, H.; Kubes, P.; Vallieres, L. Rod-Shaped monocytes patrol the brain vasculature and give rise to perivascular macrophages under the influence of proinflammatory cytokines and angiopoietin-2. J. Neurosci. 2008, 28, 10187–10199. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Lapointe, B.M.; Clark, S.R.; Zbytnuik, L.; Kubes, P. A requirement for microglial TLR4 in leukocyte recruitment into brain in response to lipopolysaccharide. J. Immunol. 2006, 177, 8103–8110. [Google Scholar] [CrossRef] [Green Version]
- Larrea, D.; Pera, M.; Gonnelli, A.; Quintana–Cabrera, R.; Akman, H.O.; Guardia-Laguarta, C.; Velasco, K.R.; Area-Gomez, E.; Dal Bello, F.; De Stefani, D.; et al. MFN2 mutations in Charcot-Marie-Tooth disease alter mitochon-dria-associated ER membrane function but do not impair bioenergetics. Hum. Mol. Genet. 2019, 28, 1782–1800. [Google Scholar] [CrossRef] [Green Version]
- Vielhaber, S.; Debska-Vielhaber, G.; Peeva, V.; Schoeler, S.; Kudin, A.P.; Minin, I.; Schreiber, S.; Dengler, R.; Kollewe, K.; Zuschratter, W.; et al. Mitofusin 2 mutations affect mitochondrial function by mitochondrial DNA de-pletion. Acta Neuropathol. 2013, 125, 245–256. [Google Scholar] [CrossRef]
- Strickland, A.V.; Rebelo, A.P.; Zhang, F.; Price, J.; Bolon, B.; Silva, J.P.; Wen, R.; Züchner, S. Characterization of the mitofusin 2 R94W mutation in a knock-in mouse model. J. Peripher. Nerv. Syst. 2014, 19, 152–164. [Google Scholar] [CrossRef]
- Cartoni, R.; Arnaud, E.; Medard, J.J.; Poirot, O.; Courvoisier, D.S.; Chrast, R.; Martinou, J.C. Expression of mi-tofusin 2(R94Q) in a transgenic mouse leads to Charcot-Marie-Tooth neuropathy type 2A. Brain 2010, 133, 1460–1469. [Google Scholar] [CrossRef] [Green Version]
- Detmer, S.A.; Velde, C.V.; Cleveland, D.W.; Chan, D.C. Hindlimb gait defects due to motor axon loss and reduced distal muscles in a transgenic mouse model of Charcot-Marie-Tooth type 2A. Hum. Mol. Genet. 2008, 17, 367–375. [Google Scholar] [CrossRef]
- Lee, J.Y.; Lee, J.H.; Koo, O.J.; Bae, H.S.; Jung, M.H.; Bae, J.H.; Hwang, W.S.; Chang, Y.J.; Lee, Y.H.; Lee, H.W.; et al. Multiple sgRNAs with overlapping sequences enhance CRISPR/Cas9-mediated knock-in efficiency. Exp. Mol. Med. 2018, 50, 16. [Google Scholar]
- Kagiava, A.; Karaiskos, C.; Richter, J.; Tryfonos, C.; Lapathitis, G.; Sargiannidou, I.; Christodoulou, C.; Kleopa, K.A. Intrathecal gene therapy in mouse models expressing CMT1X mutations. Hum. Mol. Genet. 2018, 27, 1460–1473. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; van Putten, M. Assessing functional performance in the mdx mouse model. J. Vis. Exp. 2014, 85, 51303. [Google Scholar] [CrossRef] [Green Version]
- Zielasek, J.; Martini, R.; Toyka, K.V. Functional abnormalities in P0-deficient mice resemble human hereditary neuropathies linked to P0 gene mutations. Muscle Nerve 1996, 19, 946–952. [Google Scholar] [CrossRef]
- Olympiou, M.; Sargiannidou, I.; Markoullis, K.; Karaiskos, C.; Kagiava, A.; Kyriakoudi, S.; Abrams, C.K.; Kleopa, K.A. Systemic inflammation disrupts oligodendrocyte gap junctions and induces ER stress in a model of CNS manifestations of X-linked Charcot-Marie-Tooth disease. Acta Neuropathol. Commun. 2016, 4, 95. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| sgRNA-PAM | |
|---|---|
| sgRNA #1 | ATTTGAGCAGCACACAGTCC-GGG |
| sgRNA #2 | AATTTGAGCAGCACACAGTC-CGG |
| sgRNA #3 | GACTGTGTGCTGCTCAAATT-TGG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stavropoulos, F.; Sargiannidou, I.; Potamiti, L.; Kagiava, A.; Panayiotidis, M.I.; Bae, J.H.; Yeom, S.C.; Lee, J.Y.; Kleopa, K.A. Aberrant Mitochondrial Dynamics and Exacerbated Response to Neuroinflammation in a Novel Mouse Model of CMT2A. Int. J. Mol. Sci. 2021, 22, 11569. https://doi.org/10.3390/ijms222111569
Stavropoulos F, Sargiannidou I, Potamiti L, Kagiava A, Panayiotidis MI, Bae JH, Yeom SC, Lee JY, Kleopa KA. Aberrant Mitochondrial Dynamics and Exacerbated Response to Neuroinflammation in a Novel Mouse Model of CMT2A. International Journal of Molecular Sciences. 2021; 22(21):11569. https://doi.org/10.3390/ijms222111569
Chicago/Turabian StyleStavropoulos, Filippos, Irene Sargiannidou, Louiza Potamiti, Alexia Kagiava, Mihalis I. Panayiotidis, Ji Hyun Bae, Su Cheong Yeom, Jae Young Lee, and Kleopas A. Kleopa. 2021. "Aberrant Mitochondrial Dynamics and Exacerbated Response to Neuroinflammation in a Novel Mouse Model of CMT2A" International Journal of Molecular Sciences 22, no. 21: 11569. https://doi.org/10.3390/ijms222111569
APA StyleStavropoulos, F., Sargiannidou, I., Potamiti, L., Kagiava, A., Panayiotidis, M. I., Bae, J. H., Yeom, S. C., Lee, J. Y., & Kleopa, K. A. (2021). Aberrant Mitochondrial Dynamics and Exacerbated Response to Neuroinflammation in a Novel Mouse Model of CMT2A. International Journal of Molecular Sciences, 22(21), 11569. https://doi.org/10.3390/ijms222111569